醫療器材
510 K and Quality System Regulation (QSR)
|
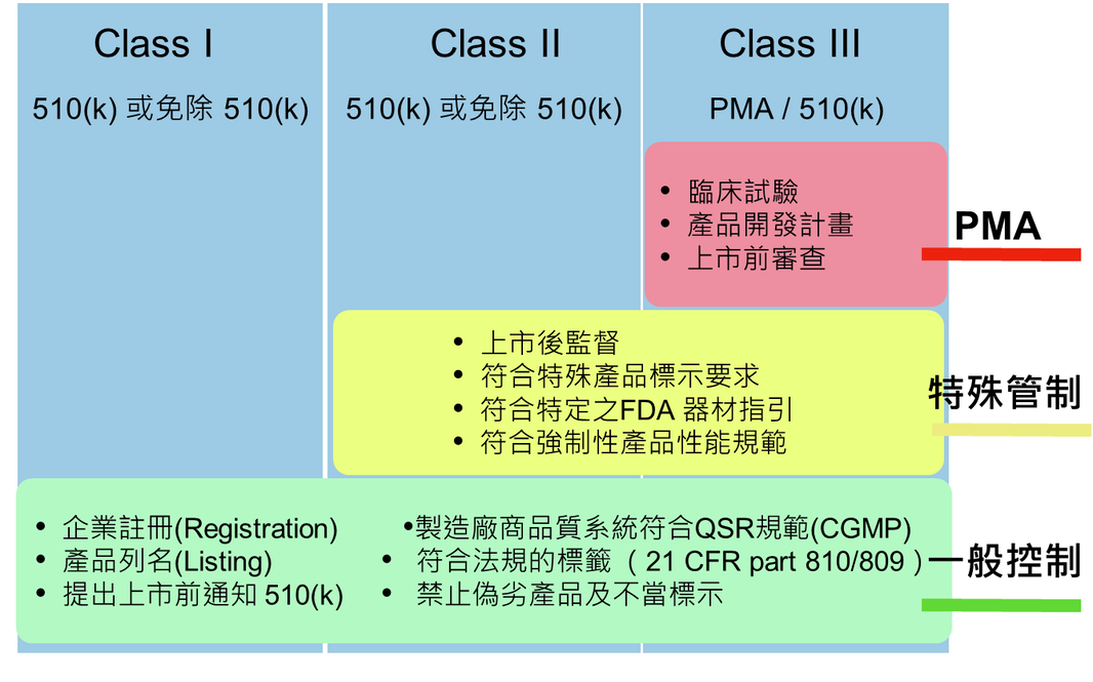
FDA 510(k): 上市前通知申請介紹: Premarket Notification PMN 行銷到美國的三級分類Class I,II,III醫療器材產品,除部分免510(k)品項及無須進行上市前核(Premarket Approval, PMA)外,都必須在進口美國前至少90天前向美國食品藥品管理局(U.S.Food and Drug Administration,簡稱FDA)提出上市前通知(Premarket Notification,PMN申請,取得輸入許可(510(k) Clearance Letter)。 Premarket Notification, 510(k):510(k)是向FDA提出的上市前相關申請文件,目的是證明與已合法上市之產品具相同安全性及有效性,此即實質相等性(substantially equivalent)。 申請人或公司必須將欲申請上市的醫療器材與已在美國FDA上市之一種或多種相似產品做比對,證明其具實質相等性。 合法上市之器材之定義為在1976年5月28日之前合法上市的器材 (Preamendment device),或者從第III類中重新分類入II或I類的器材,或者通過510(k)申請程序證明具實質相等性的器材,或者依III 類醫療器材評估定義的器材。這些醫療器材被稱為“predicate device(s)”。 綜合以上內容可知,絕大部分產品在完成企業註冊、產品列名和GMP品質系統,完成510(k)申請後,即可上市。 QSR-21 CRF 820 而所謂GMP品質系統- Quality System Regulation (QSR)即是21 CFR 820 QSR為自願性驗證: FDA不會核發QSR證書或認證。但 BEST ISO可以 發帶有 IAF LOGO13485 的QSR 驗證證書 BEST ISO 是球第一(美洲以外) 通過FDA 第三方認證法規全之第三方認證機構 QSR系統有七大部分:Management, Design controls, Production & process controls, Records, Records/documents/change controls, Material controls, Facility & equipment controls, Corrective & Preventive actions. 延伸出其他規範: Medical Device Reporting (MDR) regulation (21 CFR Part 803), Medical Device Tracking regulation (21 CFR Part 821), Corrections and Removals regulation (21 CFR Part 806) ELECTRONIC RECORDS; ELECTRONIC SIGNATURES 21 CFR Part 11- 美國21 CFR 820 -QSR醫療器材品質系統於設計、製造、包裝、標示、儲存、安裝和服務等活動所使用的方法、設施和管制。 主要是為了確保最終產品的安全及有效性,並且能夠符合美國聯邦食品藥品化妝品法之規範。 QSR提供一個框架,讓不同的醫療器材製造商,依據該發展狀況,彈性地去符合要求。 中低風險的產品進行上市前通知申請或產品列名時,FDA不會查核製造廠之QSR符合性,待產品開始於美國販售時,才查核;而高風險產品 (PMA/CLASS III 510(k))則是在上市前核可之前,就會針對製造廠品質系統進行查核,主要目的為:確認申請時所提交的生產相關資訊之正確性、評估廠商符合品質系統規範之能力。 |
聯絡我們
